Author: Columbia Biosciences
Background: Over the course of their careers, most researchers in the life sciences or associated fields will perform an experiment or assay that uses a conjugate. These include fluorescent conjugates in flow cytometry (FCS) or microscopy, enzyme conjugates for enzyme linked immunosorbent assays (ELISA), western blots (WB), or immunohistochemistry (IHC), and conjugates with metal chelate complexes for time resolved energy transfer (TR-FRET) or electrochem-iluminescence (ECL) assays. This doesn’t even include the wide array of small molecule tags and haptens such as biotin, digoxigenin, and DNP, or other biomolecules such as small peptides, nucleic acids, or bioactive drug compounds that are used as tools in all sorts of other detection schemes. What do all these conjugates have in common? They all use some form of chemistry to attach a carrier molecule to a signal generating or bridging molecule in order to find and detect a target molecule. The success with which they perform this assigned task depends greatly on how they are manufactured. This is why we’re going to discuss why researchers should understand what makes a good conjugate, well… great.
What do we mean when talking about conjugates? While conjugates can be made up of any number of different molecules or compounds, most of the ones that are used on a daily basis, and the ones we’ll be discussing here, are immunoconjugates. They are comprised of an antibody carrier that finds and binds to the target, and a detection moiety such as a fluorescent dye or enzyme, that generates a measurable signal. For these conjugates to perform successfully, both parts must be put together in a way that maintains their activity and doesn’t create any non-specific binding problems, and a good place for us to begin exploring how this can be done is by looking at the most important piece of an immunoconjugate – the antibody.
Let’s get started… look at starting materials before you leap
Antibody Quality and Purity
The ability of an antibody molecule to find and bind to a specific target is a feature that has been exploited by immunochemistry researchers for more than 60 years. As techniques to elicit, produce, and purify these antibodies have improved over the years, the possibility for creating highly sensitive and specific conjugates has followed suit. However in the end, all immunoconjugates are only as good as the antibody used to make them. Many users that are making their own conjugates or contracting that out to outside labs will source an antibody from a supplier and rely on that supplier to provide a “pure” antibody. In this scenario, what exactly does pure mean?
For the sake of simplicity, we’ll limit our discussion to monoclonals. Since the antibodies present in hybridoma cultures or ascites are often assumed to be clonal in specificity, purifying things down to an immunoglobulin only state is usually the primary focus. This is usually accomplished by using affinity columns containing Protein A or G agarose to bind up the antibody fraction while allowing other molecules to flow through and be washed away. The captured antibodies can then be eluted under low pH or high salt conditions to produce pools that are fairly uniform in specificity. However, the conditions used to elute the antibody from the solid phase can also cause the formation of higher molecular weight aggregates or low molecular weight breakdown fragments that can typically be seen using size exclusion HPLC. Many antibody suppliers might perform an SDS polyacrylamide gel (SDS-PAGE) to determine purity, but these only show if there are any other protein bands present that don’t correspond to the heavy and light chain bands, and often don’t indicate the presence of the other unwanted by-products. [FIGURE A] A secondary mode of chromatographic purification such as size exclusion can be used to clean up and remove these artifacts. If this isn’t done the unwanted components will be present during any subsequent conjugations. These can produce problems such as poorly functional conjugate, high non-specific background, and inconsistent signal in downstream assays. The purity of the starting antibody isn’t just a matter of the absence of any other proteins, but a reflection of how homogeneously monomeric the antibody itself is. In short, using a uniformly clean antibody for the conjugation will go a long way towards minimizing unwanted problems (down the line) as you proceed with your conjugation.
FIGURE A
Clean Antibody: SDS-PAGE v. HPLC

When the sample in Figure A is run on SDS-PAGE as shown in the inset (lane AMW standards, Lane B- antibody), the only bands present are those corresponding to the immunoglobulin heavy and light chains, indicating an absence of any other contaminating proteins. However, the SEC HPLC chromatogram in this case clearly shows the presence of several other peaks besides the main monomeric antibody peak. This means that while the sample is free of other non-antibody contaminants, there are unwanted aggregate and breakdown artifacts that most likely carried over from the antibody purification that should ideally be removed prior to conjugation.
Quality of Detection Moiety
Along with the antibody, the quality of the molecule that you attach to it is important as well, especially when making antibody-protein conjugates. The most common types of protein to conjugate antibodies to are enzymes, most commonly horseradish peroxidase (HRP) or alkaline phosphatase (AP), and fluorescent proteins called phycobiliproteins. It’s important to make sure that these other proteins are of a high purity to minimize variability in signal, and ensure high functionality. This is indicated by substrate turnover per unit for enzymes, and fluorescent intensity and spectral integrity for phycobiliproteins. Just like with antibodies, aggregation of these other proteins in their native form can lead to high non-specific binding and background, and low activity. Additionally, breakdown before or during conjugation of enzymes will often produce low substrate conversion and poor signal, and with phycobiliproteins will often lead to a blue-shift in spectra and poor brightness. In either case, the quality and integrity of the detection components used in your conjugates, as with the antibodies, can make a big difference when it counts at detection time.
Selection of Detection Moiety
The quality of the detection component attached to the antibody isn’t the only thing to consider when looking at conjugates. What you attach to the antibody can also have a substantial impact on how it all works. Let’s consider fluorescent conjugates as an example. Should you use small, synthetic fluorescent dyes, which are usually easier to conjugate to the antibody and come in a wide range of spectra but may not always be bright enough to generate a satisfactorily robust signal to background window? Or should you use fluorescent phycobiliproteins, which are considerably larger and can be more complex in their conjugations schemes but are usually some of the brightest fluorescent probes available and can produce extremely sensitive detection reagents?
One thing to consider is the target you’re trying to detect. Is there a lot of it present and available, like a high-density cell surface primary antigen such as CD4 or CD3, or a highly expressed recombinant outer membrane fusion protein? Or is it a low-density tertiary antigen such as CD25, or marker that is only expressed in low levels upon specific activation? When choosing conjugates for an FACS panel, highly expressed antigens can often be assigned conjugates using small fluorescent dyes, of which there are a wide variety of spectral choices to match available channels, while the phycobiliproteins such as RPE and APC can be used specifically for those targets that require higher sensitivity and brightness.
The location of the target antigen can also be a determining factor in that antibody- phycobiliprotein conjugates are by nature somewhat larger in size than those using small molecule dyes. This can be important if your target antigen is intracellular as the size of the conjugate can affect the accessibility of the target to the conjugate. While APC and PerCP conjugates, and their cyanine dye tandems, can usually be produced to be small enough to reliably access the cell interior, RPE conjugates can often be large enough to affect and limit their consistent penetration of the permeabilized cell membrane.
Unwanted attractions: Matrix effects and label interference
A less common but still occasionally relevant issue to consider is how the label you choose might itself interact with your test samples. There have been rare but documented cases in the literature of RPE conjugates specifically binding to certain subpopulations of mouse plasma cells even though the antigen targeted by the antibody is not present on those cells. RPE has also been shown in isolated cases to bind to some populations of LPS or Concanavalin A stimulated mouse B or T lymphocytes. While these cases tend to be relatively rare events, they do demonstrate the need to use the appropriate fluorochrome labeled controls to check for such occurrences. Taking a few minutes up front to think about or discuss the labeling options available for conjugation can often prevent costly errors in data interpretation later on when it’s too late.
Antibody conjugation chemistries
Common chemistries
Up to now we’ve discussed why it’s important to put some thought into what’s going into your antibody conjugate, but an equal if not more important factor is how, and to what extent, the antibody is covalently coupled with the detection molecule of choice. The most common chemistries out there utilize amine-reactive crosslinkers to target lysine side chains as points of attachment on the antibody, and in the case of enzyme or phycobiliprotein conjugates, on the other protein as well. While most antibodies have a large number of available reactive groups and it is fairly easy to incorporate a substantial number of linkage points, these reactions are stochastic in nature and can be highly dependent on the relative concentrations of both the protein and the crosslinker, as well as the reaction pH and buffer system used. Very successful conjugates are often made using these chemistries but it’s important to have a good understanding of the variables involved to prevent under conjugation which can lead to poor yields, or over conjugation which can produce unstable or poorly functional products.
Another possible factor to keep in mind when using the amine reactive strategies is that there is usually no effective way to control where the linkages are made on the antibodies. The random nature of the locations where the reactions occur can open up the possibility of steric problems during the assay. While these problems tend to be uncommon, and it’s fairly rare to come across an antibody that’s rendered nonfunctional due to a reaction site being located in the hypervariable binding region on the arms of the antibody, it has been reported and its worth remembering that every intact native antibody contains at least 4 amines, each located at the N-terminus of both of the heavy and light chains.
Fc site specific conjugations
If you’re interested in a more structurally directed conjugation, attempts to try and localize the point of attachment to the antibody were developed in the past that involved the oxidation of the carbohydrate moieties on the Fc region to generate aldehydes, which could then be reacted with amines on another crosslinker or protein to form a Schiff base, or later on with hydrazides to form hydrazone linkages, which were somewhat more stable than those between aldehydes and amines. In either case, these methods offer the possibility of conjugations that were limited to the Fc-region of the antibody, but in practice can be difficult to control and often have poor yields. As they say, your mileage may vary.
Recently, new strategies to perform site-specific conjugations have been developed that offer much greater control over how and where the detection moiety is attached to the antibody. These involve either the remodeling of the carbohydrate chains on the Fc region to incorporate specific biorthogonal reactive sites or the active labels themselves, or they utilize the high affinity binding of a peptide ligand to the Fc region, which can then be covalently linked to form a stable conjugate. These new techniques can theoretically be used to attach all different kinds of detection molecules to antibodies in a reproducible fashion. While there are still problems to be solved as with any new technology, these methods show promise in closing the gap towards making explicitly defined immunoconjugates.
Hinge region reduction
Another method of trying to control the attachment location of the detection molecule to the antibody involves another technique that has been in use for decades and is still popular, and that involves using a strong reducing agent to cleave the hinge region disulfide linkages in the antibody to try and produce two identical, monovalent heavy chain-light chain fragments. These fragments each then contain free thiols that can be reacted with maleimides or iodoacetamides on the other molecule to yield a relative stable thioether bond, that in theory is away from the antigen binding region. Although it’s a relatively simple scheme in theory, and still widely used commercially to produce successful conjugations, it’s worth remembering that your antibody is undergoing what could be considered a destructive modification, and that the hinge disulfides are not the only inter- or intra-chain disulfides that might undergo reduction. It’s always a good idea to check that the affinity of your antibody for its target antigen hasn’t been impacted significantly before putting it to work in your final assay.
How much labeling is enough labeling?
We’ve talked about the how when it comes to performing the conjugations, and now finally we need to talk about the how many. Simple logic would suggest to you that putting more of your detection molecule, whether it’s an enzyme, a fluorescent dye, a hapten, or whatever you might choose, would be better for generating a higher signal in your assay. But as is often the case, nature doesn’t always agree with this logic.
Some attachments, like many of the commercially available haptens, can in fact often be added to an antibody in fairly large numbers (6-12 per antibody) because they’re usually only serving as targets themselves for a second antibody to bind to and hopefully amplify the overall signal. However, if you try and do this using many small molecule fluorescent dyes, you find that the signal per antibody rises as you reach a fluor to protein (F/P) ratio of maybe 3-5, but then counterintuitively begins to decrease as the F/P ratio increases beyond that. [FIGURE B]
FIGURE B
Degree of conjugate labeling should be optimized for each detection moiety (label, fluorophore)

This chart shows that different fluorescent dyes can behave differently in terms of the amount of signal produced relative to the degree of labeling. In this case, Dye A produces a near linear increase in signal as the number of dyes incorporated onto the antibody increases from just over 2 to around 8. In comparison, only around 5 FITC molecules can be incorporated onto the antibody before the signal begins to be attenuated.
This phenomenon can occur for several different reasons. One simple cause is that many small molecule dyes have a relatively short Stokes shift, which is the separation between the absorption and emission maxima for a given electronic transition state. This means that there can be substantial overlap between the excitation and emission regions of the spectra for the same molecule, which can lead to destructive non radiative transfer between dye molecules. In other words, photons from your excitation source might be absorbed by a molecule of dye, exciting its electrons into a higher state, but rather than undergoing decay back to their ground state and radiatively emitting a new lower energy photon, that energy might be reabsorbed by another molecule of the same dye, repeating the process until the energy is simply dissipated as heat with no new photon emitted (known as self-quenching).
So why does this happen more at higher labeling densities? It’s most likely because as the number of dye molecules per antibody increase, the distance between any two individual molecules decreases, and since the efficiency of non-radiative transfer is highly dependent on distance, the closer the molecules are to each other, the more likely that transfer will occur, and the overall emitted signal will drop. Electronic phenomena that cause shifts in the fluorescent lifetimes of the dye can also occur in this situation. It’s also been shown that large, multi-ring, planar molecules like fluorescein can essentially “stack” when in close proximity to each other or other similar molecules. This can lead to a photoinduced electron transfer, which can then decrease the emission over time periods greater than the fluorescent lifetime itself, essentially quenching the fluorescence of the dye. The take home message for small molecule fluorescent dyes is basically not to put too few, and not to put too many, but rather find that “Goldilocks” region that will give you the biggest fluorescent bang for your buck.
The larger phycobiliprotein dyes typically don’t have quite as many issues with spatially induced problems upon conjugation, but a failure to control the reaction conditions correctly can potentially cause problems with stability and inconsistent signal response with the final conjugate. Phycobiliproteins are fairly large compared to small molecule dyes (40-250 kDa compared to 400-1500 Da), and unlike smaller dyes which tend to only have one reactive site per molecule, the proteinaceous phycobiliproteins usually have multiple reactive sites on their surfaces similar to antibodies. This means that if the reaction conditions are too aggressive with high molar excesses of crosslinker to protein at high reactant concentrations, then over conjugation can occur leading to multiple antibodies attached to multiple phycobiliproteins in one super complex that can often approach 1000 kDa or greater in size. [FIGURE C]
This hyperconjugation can lead to aggregation, either passively through hydrophobic interactions between conjugate complexes, or actively by continued reactivity of excess unreacted functional groups on the protein surfaces. This problem can be mitigated by an understanding of the stochastic nature of the reaction chemistries during crosslinking, and a careful control of stoichiometries and conditions to balance product yield with product functionality and performance. This isn’t to say that all big conjugates by definition are bad and all small conjugates are good. Some assays can potentially benefit from larger, multivalent immunoconjugates if the detection target is typically at low concentrations or surface density, and the dynamic range of the assay response doesn’t need to be too broad. However, these situations are not all that common and the parameters around them must be thought out carefully to prevent problems down the road.
FIGURE C
Amount of crosslinker offered during conjugation plays a critical role in quality of produced conjugate
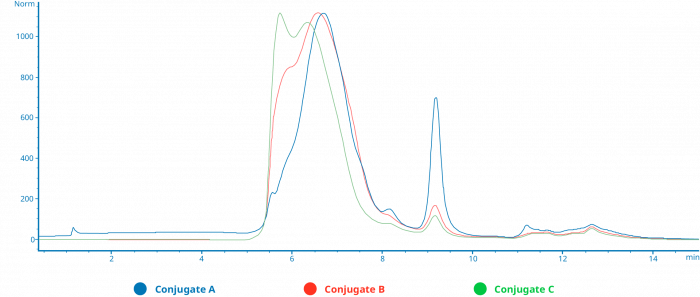
This overlay of SEC HPLC chromatograms show traces made by three different antibody enzyme conjugation reactions, where the only difference between the three was the molar excess of crosslinker offered to each protein during the individual half reactions, increasing from A to C. The molar equivalents of enzyme to antibody were kept constant across the three reactions. A clear difference in the size of the resulting products can be seen, going from a relatively uniform population of smaller conjugate species in A, to multiple populations in C, some of which approach the exclusion limit of the column.
Finishing the conjugation–purification critical
Of course, the conjugation reaction itself is only half of the problem to be solved, with the other half being the adequate purification of the final product. Most immunoconjugates are typically purified by size exclusion chromatography (SEC), often in isolation but occasionally in combination with another mode such as ion exchange (IEX) or hydrophobic interaction chromatography (HIC). When the size difference between individual conjugate populations in a reaction gets too small to be resolved adequately by SEC, IEX can often be used to separate groups based on the charge difference between the antibody and the other protein, and the overall aggregate charge of the complex as a whole. HIC can be used to separate complexes based on their overall hydrophobicity, which can prove useful in stripping out populations of conjugate whose increased hydrophobic nature might lead to later problems with aggregation.
Downstream analysis of FPLC purified conjugate fractions using SEC-HPLC is a valuable tool in characterizing population characteristics. Measuring an F/P ratio by UV-Vis spectroscopy alone can occasionally be misleading because it’s generating a value by measuring the bulk sample as a whole, fractions or pools that still contain mixed populations of conjugate complexes will often reflect the overall empiric F/P ratio- i.e., what appears to be a 1:1 ratio might actually consist of a mixture of 1:1, 2:2, or 3:3 complexes, all of which would have the same basic spectrophoto-metric ratio. It’s the observation of how the different samples might show shifts in peak retention times on a higher resolution SEC-HPLC column that can help you illuminate the subtle differences in composition. [FIGURE D]
Figure D
SEC HPLC analysis must be included into final assessment of conjugate quality
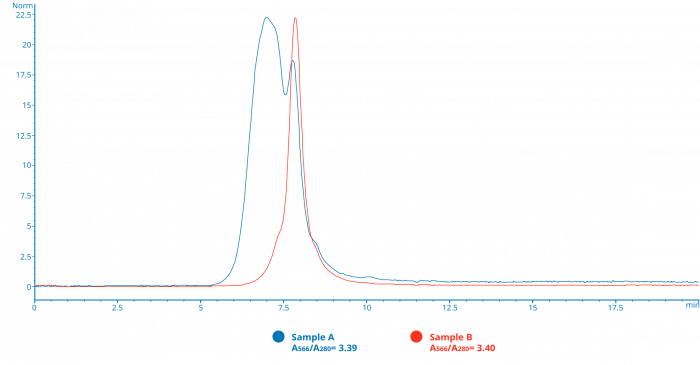
This SEC chromatogram shows traces of two separate pools of fractions purified from an antibody-R-phycoerythrin conjugation. (Samples A and B) Both pools have nearly identical ratios of absorbances at 566nm to 280nm, a value often used to calculation the final Fluor to Protein ratio (F/P), which if viewed in isolation would seem to indicate that the populations in both samples have a similar degree of conjugation and final composition. However, the chromatograms clearly show that Sample A has a much larger amount of higher order conjugate populations than B, and as such might perform quite differently in the final assay tested.
Other tools such as online chromatographic light scattering (DLC or MALS), SDS-PAGE, or activity-based testing of fractions can also be used to further help characterize the composition of conjugates. When working with antibody protein conjugates, the size of the desired final product is often influenced by the type of application that it will be used in. Many flow cytometry fluorescent conjugates are often relatively small in size, often striving to be as close as possible to the widely sought after 1:1 F/P ratio. This small conjugate size helps maintain a higher degree of homogeneity in the final product, which can improve signal by minimizing steric problems, often leads to tighter and more uniform staining of targeted cell populations.
Many antibody-enzyme conjugations for ELISA or western blot applications, on the other hand, can sometimes be designed to produce a larger and more multimeric product, one in which the antibody component has a higher degree of multi-valency and avidity, and more enzymes can be delivered to the target site to turn over as much substrate as possible. Of course, the same cautionary tales that we discussed several times earlier apply equally here, and in short, you as the researcher and end user need to be careful with what you wish for. Conjugates, like clothes, are a good example of how the “one size fits all” philosophy does not always apply as often as we might like it to.
About Columbia Biosciences – we’re here to help
As detailed as our discussion of the ins and outs of what makes a “great” conjugate seems, this is by no means an exhaustive look at the subject. This can pose a problem for researchers who know they need a conjugate for a particular assay application but may not know quite where to start. This is a good example of why it can be helpful to speak to someone who has experience in these situations, and Columbia Biosciences can provide that level of expertise in helping you navigate your way through the process. Whether you have years of experience working with or even making your own conjugates, or you weren’t even aware of what a conjugate was before reading this, our conjugation experts can talk with you about your needs and requirements and come up with a plan to provide you with a conjugate that can be as broadly or specifically focused as necessary.
We have years of experience with all different types of crosslinking chemistries, from the classic strategies applied to thousands of different conjugates commercially or otherwise, to the newest site-directed schemes designed to produce more uniform and consistent reagents than ever. We also have access to and experience with a wide range of labels (enzymes, small molecule tags, and metal chelates, to all different types of fluorescent dyes including the phycobiliproteins) for which we are one of the few conjugation houses that actually produce our own native phycobiliproteins under an ISO 13485:2016 quality management system to ensure a consistent high-quality supply.
Our conjugation scale ranges from sub-milligram to multi-hundreds of milligrams, each performed with detailed batch records and testing to ensure batch to batch reproducibility. We currently perform custom conjugations involving antibodies or other proteins which are going into pre-IVD or pre-clinical applications, and most are completed with an average two-week turnaround time. Columbia also has experience with and performs other types of bioconjugation services involving peptides, nucleic acids, PEGs/polymers, carbohydrate, drugs, beads, and more specialized antibody drug conjugates (ADCs). As such we count among our customers numerous large and small pharma companies, established to startup biotech organizations, and CROs working in everything from basic early-stage reagent and assay development to the execution and administration of ongoing therapeutic clinical trials. In the end, no customer is too big or too small for us to help succeed.
We also have the capability of going beyond just the conjugation phase of reagent development and have the experience and capabilities to do downstream assay development in applications such as HTRF/ TR-FRET for compound library screening, flow cytometry and other cell-based assays, custom cell line development, and many other areas needed by our customers. Our analytical capabilities consist of most of the industry standards such as HPLC using several different detection systems, UV-Vis spectroscopy, SDS-PAGE/western blotting, and ELISA, but also include in-house flow cytometry testing, Luminex assays, TR FRET assays, small molecule degree of labeling (DOL) using mass spectrometry, and surface plasmon resonance (SPR). As well as the conjugation service itself, we can also work with our customers who have specialized formulation requirements such as unusual buffer systems or multi-vial aliquoting and lyophilization.
In conclusion, Columbia Biosciences has the proven experience and capability to help walk you the researcher through the complex arena of reagent development. We can go beyond showing you what is simply a good conjugate, and instead help you focus on finding the best conjugate to ensure that your projects, however big or small, complex or simple, will be as successful as they can be. Get in touch with us today to talk with some of our conjugation professionals and let us show what we can do for you.
Click here to access the PDF version of this whitepaper from Columbia Biosciences.




